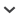The remaining populations of ocelot (Leopardus pardalis albescens) in the United States are reduced to 2 isolated populations in southern Texas, with the next closest populations occurring in central Tamaulipas, Mexico. The species is listed as endangered, and recovery of populations in Texas eventually might require translocations from larger source populations. We sequenced the mitochondrial DNA control region from individuals from Texas and northern Mexico and compared these data to existing sequences derived from ocelots in other parts of its range in southern Mexico, Central America, and South America. Nucleotide diversity was lower in Texas than in Mexico, suggesting a loss of genetic variation as a consequence of fragmentation and increased genetic drift. Phylogenetic analyses showed a close relationship between populations in Texas and northern Mexico that encompass the range of the subspecies L. pardalis albescens. Based on these data, the best source population for a recovery plan involving ocelot translocations would be northern Mexico, because this region seems to form a discrete management unit (both ecologically and phylogenetically) that includes Texas.
The historical range of the ocelot (Leopardus pardalis) once extended from South America into parts of the United States (Guggisberg, 1975; Hall, 1981; Navarro et al., 1993). Habitat degradation, human encroachment, and uncontrolled harvest in the 1800s and early 1900s extirpated the ocelot from most of its range in the United States, with current populations confined to 2 areas in southern Texas (Koford, 1978; Tewes and Everett, 1986; Navarro et al., 1993). Ocelots in southern Texas and the northern Mexican state of Tamaulipas occupy the Tamaulipan Biotic Province and represent the subspecies L. p. albescens. In this region, ocelots exhibit strong habitat selection for dense, native thorn-shrub communities with >95% canopy cover, characteristic of the Matamoran District (Tewes, 1986; Murray and Gardner, 1997). However, >95% of these communities in southern Texas and northern Mexico have been removed (Navarro, 1985; Tewes, 1986; Tewes and Everett, 1986; Jahrsdoerfer and Leslie, 1988; Laack, 1991; Caso, 1995; Shindle and Tewes, 1998). In the Tamaulipan Biotic Province, the remaining habitat used by ocelots occurs in small, fragmented patches surrounded by urbanization, agriculture, and other human developments (Navarro et al., 1993). Although ocelots still are considered abundant in parts of Mexico, Central America, and South America, only 80 to 120 individuals are subdivided into the 2 remaining populations in southern Texas (Tewes and Everett, 1986). Today these 2 isolated populations are listed as endangered under the U.S. Endangered Species Act (U.S. Fish and Wildlife Service, 1999), and the subspecies L. p. albescens is listed internationally as Appendix I by CITES (2005).
Genetic data from microsatellite loci (Walker, 1997) indicated less heterozygosity in ocelot populations occupying southern Texas relative to those occurring in northern Mexico. This loss of variation is presumably the consequence of recent range fragmentation, reduced effective population sizes, and increased genetic drift (Walker, 1997). The genetic consequences of this fragmentation are especially pronounced in the population at Laguna Atascosa National Wildlife Refuge (Laguna Atascosa NWR). One proposed method for recovering the loss of genetic variation in the Texan populations involves the translocation of individuals from another source population with a higher level of genetic variation (Brook et al., 2002). Selecting an appropriate source population is important for offsetting any potential deleterious effects from mixing genetically divergent populations (Storfer, 1999; Edmands and Timmerman, 2004; Goldberg et al., 2005). Although populations in southern Texas and northern Mexico are placed in the same subspecies, their phylogenetic position relative to each other as well as to other populations is unknown (Murray and Gardner, 1997). A recent molecular phylogenetic study of subspecies in primarily Central and South America indicated a lack of congruence between currently recognized boundaries of the 10 subspecies of ocelots and groups identified with genetic data (Eizirik et al., 1998). Therefore, the validity of recognized subspecies of ocelot is unclear.
We had 2 primary objectives. First, we examined mitochondrial DNA (mtDNA) variation in the control region from ocelots in southern Texas and northern Mexico to evaluate existing levels of variation. Second, we derived a molecular phylogeny from mtDNA sequences to evaluate the most appropriate source populations for recovery efforts involving translocations and to determine the phylogenetic placement of ocelots from southern Texas and northern Mexico.
Methods
Blood samples were obtained from wild-caught ocelots (n = 9) captured during previous radio-telemetry studies at 2 areas in southern Texas and one in northern Mexico (Laack, 1991; Caso, 1995; Shindle and Tewes, 1998). All 3 areas were within the Tamaulipan Biotic Province (Blair, 1950). Specific sampling localities included: Laguna Atascosa NWR, Cameron County, Texas (n = 1); private ranches in northern Willacy County, Texas (n = 5); and private ranches in southern Tamaulipas, Mexico (n = 3) (Fig. 1). Ocelots were captured, immobilized, and processed using methods developed by Tewes (1986) and Beltran and Tewes (1995). Approximately 3 cc of blood were taken from each captured individual and maintained in Longmire's lysis buffer (Longmire et al., 1997). We realize that the sample sizes are small, but they are equivalent to the number of individuals previously examined by Eizirik et al. (1998) for ocelots in southern Mexico, Central America, and South America. Therefore, samples collected by us from the range of L. p. albescens allowed an evaluation of relationships among populations in northern Mexico and southern Texas relative to other areas of Latin America.
Figure 1
Map showing approximate locations where ocelot (Leopardus pardalis) samples were obtained, with the approximate historical distributions of ocelot subspecies symbolized by shading.
Designations of subspecies (Oliveira, 1994; Murray and Gardner, 1997) are in parentheses: L. p. sonoriensis (son); L. p. albescens (alb); L. p. pardalis (par); L. p. nelsoni (nel); L. p. aequatorialis (aeq); L. p. pusaea (pus); L. p. pseudopardalis (pse); L. p. melanura (mel); L. p. mitis (mit); L. p. steinbachi (ste). Locations in the range of L. p. albescens (denoted TX for Texas and TAM for Tamaulipas in northern Mexico) were sampled in this study, and the other locations were sampled by Eizirik et al. (1998).

Blood samples were stored in Longmire's solution (Longmire et al., 1997), and total genomic DNA was isolated following Seutin et al. (1991). The extracted DNA was used as a template for polymerase chain reaction (PCR) amplification and sequencing of an approximately 1,179 bp fragment from the mitochondrial control region. Nine individuals were examined from the Tamaulipan Biotic Province. The mitochondrial control region and adjacent tRNA genes were amplified using primers L16215 (5′-TACACTGGTCTTGTAAACC-3′) and H938 (5′-AAGGCTAGGACCAAACCT-3′). Primers from the heavy (H) and light (L) strand were numbered according to the domestic cat sequence (Lopez et al., 1996). In addition to these external primers, 7 internal primers were used for sequencing: L16391 5′-TGTGCTTGCCCAGTATGTC-3′; L16897 5′-CTCTTCTCGCTCCGGGCCCA-3′; L129 5′-CTTGTAGCTGGACTTATT-3′; H727 5′-ATGACAGGGGATTGGTAAAGC-3′; H41 5′-AAAATACCAAATGCATGACA-3′; H228 5′-CATGCCGTGTCCTGTGGAAC-3′; and H16873 5′-CCGGAGCGAGAAGAGGTAC-3′. Amplification was performed in 20-µL PCR reactions containing 25 ng genomic DNA, 2.5 mM MgCl2, 50 mM Tris-HCl, 0.30 mM of each dNTP, 0.1 µL BSA, 0.15 U Taq polymerase (Applied Biosystems, Foster City, California), and 1.0 pmol of each primer. Conditions for PCR amplification included an initial denaturation at 96°C for 3 min, followed by 32 cycles with a denaturation at 93°C for 30 s, re-annealing at 50°C for 1 min, and an extension at 72°C for 1 min, with a final extension for 10 min at 72°C. Amplification products were purified using the Prep-a-Gene DNA purification kit (Bio-Rad, Hercules, California). An aliquot of the PCR product was ligated into pBluescript plasmid (Stratagene, Kirkland, Washington), which was modified to contain thymine overhangs at the 3′ end by digestion with EcoRV and incubation with Taq polymerase (Applied Biosystems) and dTTP (Alting-Mees and Short, 1989). Ligation products were transformed using competent DH5α cells grown on Luria-Bertani (LB) plates containing ampicillin, isopropylthiogalactoside, and X-gal. DNA was extracted from 100-mL aliquots of cell cultures derived from isolated positive colonies (Sambrook et al., 1989). Phenol/chloroform/isoamyl alcohol (24∶1∶1) extraction was used to partially purify the plasmid DNA, and insert length was verified by electrophoresis on a 0.8% agarose gel with 10× glucose, stained with ethidium bromide and visualized under UV light. Colonies with plasmid inserts of approximately 2,000 bp were sequenced using the dideoxy chain termination method, α-35S-dATP for labeling, a combination of primers and gradient gel electrophoresis (Biggins et al., 1983). We exposed sequence gels for 12 to 24 h on Kodak Bio-Max film and sequenced 2 or more clones for each individual to verify sequences.
We estimated diversity of haplotypes and nucleotides with the DnaSP program (Rozas and Rozas, 1999). Phylogenetic comparisons of ocelots from the Tamaulipan Biotic Province to those from other regions of Central and South America were restricted to the 410 bp control region fragment previously reported by Eizirik et al. (1998). Control region sequences from Eizirik et al. (1998; locations in Fig. 1) were downloaded from GenBank and aligned with control region sequences obtained in this study. Alignment was performed in CLUSTAL X (Thompson et al., 1997). Phylogenetic analyses were performed using maximum parsimony (MP) and maximum likelihood (ML) in PAUP* (Swofford, 2001). Maximum-parsimony analysis employed the heuristic search option with TBR branch swapping and 100 random additions. Support for clades was determined using the bootstrap method with 1,000 replications and the same heuristic search option. Before performing the ML analysis, we selected the appropriate model (HKY+I+G) using ModelTest version 3.06 (Posada and Crandall, 1998). Bootstrap values for the ML tree were estimated using the fast version in PAUP* with 1,000 replications. Outgroup taxa for both MP and ML analyses consisted of 2 sequences from margay (Leopardus weidii) (Ezirik et al., 1998).
Results
Genetic Diversity
Sequences of the control region from ocelots revealed 2 areas, RS-2 and RS-3, containing tandem repeats that produced size heteroplasmy. We excluded these 2 regions from further analysis. A 1,179 bp fragment from the control region was sequenced and aligned for 9 ocelots from Texas and northern Mexico. This region revealed 9 variable sites distributed among 5 haplotypes (Table 1). Two haplotypes were identified among the 6 ocelots from Texas, and all 3 of the ocelots from Mexico represented different haplotypes. Haplotype diversity in Texas was 0.33 compared to 1.00 in Mexico; nucleotide diversity also was higher in the Mexican sample, which was collected from one area (Table 1). For the 410 bp control region fragment, 4 haplotypes were observed for ocelots from the Tamaulipan Biotic Province, and relative to populations in Central and South America, these 4 haplotypes had considerably lower nucleotide diversity (Table 1).
Table 1
Number of haplotypes, number of variable sites, nucleotide diversity, and mean number of nucleotide differences per site among 35 ocelots (Leopardus pardalis) originating in North, Central, and South America. We sequenced haplotypes from the Tamaulipas Biotic Province, Mexico, and obtained sequences of ocelots from other regions from Eizirik et al. (1998).

Phylogenetic Analysis
Haplotype phylogenies produced by MP and ML were congruent in several respects (Figs. 2, 3). Both revealed a clade containing haplotypes from Texas, Mexico, and most Central American localities with the exception of Panama. The 4 haplotypes found in the Tamaulipan Biotic Province (Texas and northern Mexico), representing the subspecies L. p. albescens, formed a monophyletic group most closely aligned with a haplotype from Nicaragua (Lpa25; Eizirik et al., 1998). The other group within this major clade contained haplotypes found primarily within the range of L. p. pardalis. Although Nicaragua is within the range of L. p. aequatorialis, the haplotype identified by Eizirik et al. (1998) shared with other members of this clade a 7 bp deletion not found in haplotypes from Panama and South America. The second largest clade contained haplotypes from localities in Panama and portions of South America. Relationships among these haplotypes were not resolved well. With few exceptions (such as L. p. pseudopardalis), most haplotypes did not group according to subspecific boundaries. The MP and ML analyses disagreed with respect to whether or not all South American haplotypes formed a monophyletic group, and part of this disagreement related to the placement of haplotypes from French Guyana and parts of Brazil (Figs. 2, 3). In addition, they disagreed over how particular groups within the larger South American clade were defined.
Figure 2
Phylogenetic relationships among ocelot (Leopardus pardalis) haplotypes based on a 410 bp portion of mtDNA control region.
Ocelots marked with # were from the Tamaulipas Biotic Province and were sequenced for this study; all other sequences were obtained from Eizirik et al. (1998). The phylogeny was constructed using the MP method in PAUP* (length = 252, CI = 0.456, trees with identical score = 7), based on a consensus of 1,000 bootstrap replicates, and bootstrap values >50 are shown.

Figure 3
Phylogenetic relationships among haplotypes based on a 410 bp portion of the mtDNA control region. Ocelots (Leopardus pardalis) marked with # were from the Tamaulipas Biotic Province and were sequenced in this study; all other sequences were obtained from Eizirik et al. (1998). The phylogeny was constructed using the ML method in PAUP* (length = 160, CI = 0.545). ModelTest was used to select the most appropriate mutation model (Posada and Crandall, 1998). The tree was based on the HKY+I+G model, with 0.3145, 0.2743, 0.1348, and 0.2764 (A, C, G, T, respectively), frequencies, Ti/Tv ratio of 8.2287, 0.4904 proportion of invariable sites, and 0.5538 gamma distribution. Bootstrap values were obtained using 1,000 bootstrap replicates by using the fast bootstrap implemented in PAUP*.

Discussion
Based on an examination of 10 microsatellite loci, Walker (1997) reported a reduction in mean heterozygosity in populations of ocelots from Texas compared to those in Mexico, with heterozygosity of the isolated population at Laguna Atascosa NWR being approximately half that seen in Mexico. In addition, an assessment of single-strand conformation polymorphism (SSCP) for a fragment of the mitochondrial cytochrome b gene revealed 2 haplotypes in the Mexican population (Walker, 1997). The population at Laguna Atascosa NWR was fixed for the low-frequency cytochrome b haplotype observed in the Mexican population, and the predominant haplotype observed in the other population from Texas was the same high-frequency haplotype seen in Mexico (Walker, 1997). Estimates of haplotype diversity and nucleotide diversity, derived from the entire control region sequence as well as the 410 bp fragment, were lower in Texas than in populations from Mexico, thus supporting the suggestion by Walker (1997) that populations in Texas are experiencing a decline in genetic variation relative to populations in Mexico and elsewhere.
The ocelot has been divided historically into 10 subspecies (Murray and Gardner, 1997). As revealed earlier by Eizirik et al. (1998), in most cases the mtDNA phylogeny is not congruent with the range of recognized subspecies (Figs. 2, 3). Nevertheless, there is evidence for 2 major geographical units, one in southern Texas, Mexico, and northern Central America, and the other in southern Central America and South America. Each major clade shows some evidence of substructure; for example, the Texan populations are most similar to those in northern Mexico, thus defining a unit in the Tamaulipan Biotic Province within the range of L. p. albescens. A similar pattern has been observed for the ferruginous pygmy-owl (Glaucidium brasilianum), a species that has been reduced to small fragmented populations in southern Texas that are adjacent to larger populations in Mexico and Central America (Proudfoot et al., 2006). Texas populations of pygmy-owl are represented by one mtDNA haplotype, which appears closest to haplotypes in Tamaulipas, Mexico. In addition, pygmy-owls in the Tamaulipan Biotic Province seem to represent a distinct unit from more southern populations in Mexico. The distinction of populations of ocelot and pygmy-owl in the Tamaulipan Biotic Province might represent historical events that occurred during the Pleistocene.
Human activities in southern Texas have eliminated large tracts of dense thorn-shrub communities preferred by ocelots (Tewes and Everett, 1986; Jahrsdoerfer and Leslie, 1988). This has resulted in the fragmentation of ocelot populations in the Tamaulipan Biotic Province and significant genetic erosion in the Texan populations (Thorton, 1977; Navarro, 1985; Tewes, 1986; Tewes and Everett, 1986; Walker, 1997). This loss of genetic variation likely is to continue without some type of intervention (Walker, 1997). One potential method of either sustaining or increasing levels of genetic variation in populations in Texas, thereby ensuring long-term population viability, is to augment existing populations with translocations from Latin American populations (Brook et al., 2002). Based on genetic data from Walker (1997) and from our study, populations in the Tamaulipan Biotic Province seem to be part of a more widespread population that once included northern Mexico and encompassed the range of L. p. albescens. Currently, the 2 isolated populations in Texas are not managed as a single unit. Given the apparent erosion of genetic variation in these 2 populations, translocations are necessary to sustain genetic viability. Because populations in Texas and northern Mexico are genetically similar, regions in northern Mexico are the most appropriate source for translocations.
Acknowledgments
This study was funded by Rob and Bessie Welder Wildlife Foundation, U. S. Fish and Wildlife Department, and F. Yturria. We thank F. Yturria for access to his ranch. Hernán López-Fernández graciously assisted with the Spanish translation of the abstract. This article represents publication numbers 656 for the Rob and Bessie Welder Foundation, 06-101 for the Caesar Kleberg Wildlife Research Institute, and 142 for the Center for Biosystematics and Biodiversity at Texas A&M University.